
In This Issue
Longevity
-
 Loneliness as a Vital Sign: What 14 Years of UK Data Reveal About Staying Independent
A long-running English study suggests that persistent loneliness — not the occasional lonely week — quietly raises the odds of functional decline and earlier death. The signal is moderate, but it deserves a place alongside blood pressure and grip strength.
Loneliness as a Vital Sign: What 14 Years of UK Data Reveal About Staying Independent
A long-running English study suggests that persistent loneliness — not the occasional lonely week — quietly raises the odds of functional decline and earlier death. The signal is moderate, but it deserves a place alongside blood pressure and grip strength.
-
 Are Life Expectancy Gains Finally Slowing? What New Cohort Forecasts Mean for Your Healthspan
A sweeping new analysis across 23 high-income countries says the steady climb in lifespan is losing steam. Here's what that actually means for the choices you make now.
Are Life Expectancy Gains Finally Slowing? What New Cohort Forecasts Mean for Your Healthspan
A sweeping new analysis across 23 high-income countries says the steady climb in lifespan is losing steam. Here's what that actually means for the choices you make now.
-
 Reversing the Decline: What the New Science Says About Aging Muscle
A 2025 review reframes sarcopenia as a modifiable condition — and grades which interventions actually move the needle on muscle mass, fiber loss, and motor unit decline.
Reversing the Decline: What the New Science Says About Aging Muscle
A 2025 review reframes sarcopenia as a modifiable condition — and grades which interventions actually move the needle on muscle mass, fiber loss, and motor unit decline.
-
 The Sarcopenia Signal: Why Your Nerves, Not Just Your Muscles, Drive Age-Related Decline
A new systematic review reframes age-related strength loss as a problem of failing wires, not shrinking cables — and it changes what screening and training after 60 should look like.
The Sarcopenia Signal: Why Your Nerves, Not Just Your Muscles, Drive Age-Related Decline
A new systematic review reframes age-related strength loss as a problem of failing wires, not shrinking cables — and it changes what screening and training after 60 should look like.
-
 Menopause Timing May Be an Early Warning Light for Brain Aging
A large multiomic study links later menopause with healthier prefrontal cortex aging decades on — a finding worth knowing for the women in your life, and for what it suggests about how the body ages as one system.
Menopause Timing May Be an Early Warning Light for Brain Aging
A large multiomic study links later menopause with healthier prefrontal cortex aging decades on — a finding worth knowing for the women in your life, and for what it suggests about how the body ages as one system.
Medical Research
-
 Why Your Flu Shot Works Less Well at 70: The New Science of Fixing Vaccines for Older Immune Systems
Aging immune systems mount weaker responses to vaccines. A new Nature Aging review maps the engineering — higher doses, smarter adjuvants, mRNA — designed to close the gap.
Why Your Flu Shot Works Less Well at 70: The New Science of Fixing Vaccines for Older Immune Systems
Aging immune systems mount weaker responses to vaccines. A new Nature Aging review maps the engineering — higher doses, smarter adjuvants, mRNA — designed to close the gap.
-
 Inheriting Resilience: Could a Parent's Mitochondrial Stress Make You Live Longer?
A new worm study suggests that mild stress in one generation can leave longevity marks on the next. The mechanism is fascinating — and the leap to humans is still a long one.
Inheriting Resilience: Could a Parent's Mitochondrial Stress Make You Live Longer?
A new worm study suggests that mild stress in one generation can leave longevity marks on the next. The mechanism is fascinating — and the leap to humans is still a long one.


Loneliness as a Vital Sign: What 14 Years of UK Data Reveal About Staying Independent
A long-running English study suggests that persistent loneliness — not the occasional lonely week — quietly raises the odds of functional decline and earlier death. The signal is moderate, but it deserves a place alongside blood pressure and grip strength.
For most of my working life, the markers of a good checkup were predictable: blood pressure, cholesterol, a squeeze of the hand to gauge grip, a few questions about sleep. Nobody asked whether you had anyone to call on a Sunday. A new analysis out of England suggests they probably should. Researchers tracking more than five thousand older adults for fourteen years report that the people who were lonely not just once, but repeatedly across years, were measurably more likely to lose function in daily life and to die sooner than peers who weren't. The finding isn't a thunderclap. It's something quieter, and arguably more useful: confirmation that social health behaves like a vital sign, with a dose and a duration that matter.
- Chronicity is the key variable. One bad stretch of loneliness is not the same risk as years of it.
- Functional impairment rose meaningfully in adults with chronic loneliness compared with peers who weren't lonely, after accounting for other factors.
- Social isolation and loneliness are not the same thing. One is the count of contacts; the other is how it feels.
- The evidence is observational — strong enough to act on personally, not strong enough to call causal.
- Practical response: treat your social calendar with the seriousness you'd give a cardiology appointment.
What the study actually measured
The work, published in Nature Mental Health, draws on the English Longitudinal Study of Ageing — ELSA, to its friends — a panel that has followed thousands of older adults in England since 2002. Investigators Qian Gao, Andrew Steptoe and Daisy Fancourt looked at waves 2 through 9, covering 2004 to 2018, and did something most prior research hasn't: they measured loneliness and isolation repeatedly, then sorted participants into three groups — not present, fluctuating, or chronic — based on a four-year window. Then they watched what happened over the next decade. The analysis included 5,131 adults in the mortality cohort and 4,279 who were free of functional disability at baseline. Mean age was about 67. Median follow-up was just under ten years. The headline result: compared with people who weren't lonely, those with chronic loneliness had a sub-hazard ratio of 1.58 (95% CI 1.12–2.23) for new functional impairment, and chronic social isolation carried a sub-hazard ratio of 1.41 (1.02–1.94). Even fluctuating loneliness raised the risk modestly (sHR 1.30, 1.03–1.63). The confidence intervals are wide-ish but they do not cross 1 — meaning the effect is statistically present, though the precise size is uncertain.
That distinction between fluctuating and chronic is the part to underline. A lot of older men go through stretches — a spouse's illness, a friend's funeral, a long winter — where loneliness spikes and then recedes. That pattern, this analysis suggests, is not benign, but it is meaningfully less hazardous than loneliness that simply takes up residence and stays.

Researchers distinguished between social isolation — the structural count of contacts — and loneliness, the felt sense of not having them. Both mattered. Loneliness mattered more.
Why this is different from the usual loneliness headline
You have probably read, more than once, that loneliness is "as bad as smoking fifteen cigarettes a day." That line, repeated until it lost its meaning, came from a single 2010 meta-analysis and rests on a snapshot view of social connection. What the ELSA team add is the time dimension. By measuring loneliness across three successive waves before counting outcomes, they could ask a sharper question: is it the state of being lonely on a given afternoon that matters, or the trajectory? Their answer points to trajectory. People whose loneliness came and went carried less risk than people for whom it was a steady companion.
That has practical consequences. It means a difficult year, however painful, is not a sentence. It also means the goal of social-health work — for individuals, for clinicians, for the public-health apparatus — is to prevent loneliness from settling in for the long haul. Catching it while it's still fluctuating is, by this evidence, the window that matters most.
A difficult year is not a sentence. Loneliness that settles in for the long haul is the pattern to interrupt.
What the evidence will and won't carry
A word on the rating. PinnacleLife calls this evidence moderate, and the label fits. ELSA is a serious, well-run cohort, the sample is large, and the statistical approach — Cox proportional hazards for mortality, Fine-Gray competing-risk modeling for functional impairment — is appropriate for the question being asked. But this is observational work. It cannot, on its own, prove that loneliness causes decline. Lonely people differ from non-lonely people in ways that are hard to fully adjust for: prior depression, less robust health to begin with, fewer resources, narrower social networks built up over a lifetime. The authors controlled for what they could, and the association survived. That is meaningful. It is not a randomized trial.
The honest reading is this: persistent loneliness keeps company with worse outcomes in older adults, the relationship is consistent with a long line of prior research, and it would be unwise to wave it off. It would also be unwise to claim, on the strength of one paper, that fixing loneliness adds years to life in any guaranteed way. Both things are true.

The social calendar as preventive medicine: not a cure, but a hedge worth taking seriously.
What a sensible reader does with this
I am not your doctor and this column is not a prescription. But the practical takeaway from the ELSA analysis does not require a clinical degree. If you are in your sixties or beyond and you notice that loneliness has stopped being a visitor and started being a tenant — that the quiet in the house is no longer occasional but constant — that is worth treating as a clinical signal, not a character flaw. Mention it to your GP. Mention it to the people who love you. The interventions are mostly unglamorous: a standing weekly coffee, a volunteer shift, a class, a walking group, a phone call you make on a schedule rather than waiting to feel like it. None of these are tested in trials the way a statin is. All of them are within reach.
The reason to take this seriously, for the reader of this magazine, is the same reason to take resistance training and blood-pressure control seriously: independence. The outcome the ELSA team measured wasn't an abstract one. Functional impairment is the difference between dressing yourself and needing help, between climbing your own stairs and not. Anything credibly associated with that outcome belongs on the list.
The bottom line
Treat the ELSA findings the way you'd treat a credible early-warning light on the dashboard. The signal is real, the size is plausible, the mechanism is debated but unsurprising. The fix is not pharmacological and it is not fast. It is the patient rebuilding of a life with other people in it — done a little earlier, and a little more deliberately, than most of us are inclined to.
Frequently asked questions
Is feeling lonely for one difficult year as risky as being lonely for many years?
No — the analysis found that chronicity is the key variable. People whose loneliness fluctuated carried meaningfully less risk than those for whom it became a steady, persistent state, leading the authors to describe a difficult year as 'not a sentence.'
What is the difference between loneliness and social isolation in this research?
The researchers treated them as distinct: social isolation is the structural count of contacts a person has, while loneliness is the felt sense of not having them. Both were associated with worse outcomes, but loneliness mattered more.
What specific outcomes did the study track, and over how long?
Investigators followed 5,131 older adults with a median follow-up of just under ten years, watching for new functional impairment — defined as difficulty with daily tasks like dressing or climbing stairs — and mortality. The full ELSA panel spanned 2004 to 2018.
Why does the article stop short of saying loneliness causes decline?
Because the study is observational, not a randomized trial. Lonely people differ from non-lonely people in hard-to-fully-adjust-for ways — prior depression, less robust baseline health, fewer resources — and while the association survived statistical controls, the design cannot on its own prove causation.
What practical responses does the article suggest for someone who recognizes chronic loneliness in themselves?
The article recommends treating it as a clinical signal rather than a character flaw, mentioning it to a GP or loved ones, and adopting unglamorous but reachable habits: a standing weekly coffee, a volunteer shift, a class, a walking group, or a phone call made on a schedule rather than waiting to feel like it.
Sources

Are Life Expectancy Gains Finally Slowing? What New Cohort Forecasts Mean for Your Healthspan
A sweeping new analysis across 23 high-income countries says the steady climb in lifespan is losing steam. Here's what that actually means for the choices you make now.
Here's the question I keep asking lately: is the long, lucky streak of each generation living a little longer than the last finally running out of gas? For most of the last century, life expectancy in wealthy countries climbed like a stock chart your grandparents would brag about. But a new analysis suggests the line is starting to bend — gently, but clearly. And honestly? That changes how I think about my own daily choices.
Researchers writing in the Proceedings of the National Academy of Sciences just pulled off something pretty ambitious. They took multiple established (and some brand-new) mortality forecasting methods, pointed them at 23 high-income countries, and asked a simple question about everyone born between 1939 and 2000: how long are these cohorts actually going to live, on average? The methods disagreed on lots of details. But they agreed on the headline — and that agreement is the part that made me sit up.
Across the board, the forecasts indicate a deceleration in cohort life expectancy. Translation, in plain English: the engine that's been adding extra years to each generation is downshifting.
Wait — what's a "cohort," exactly?
Quick gloss, because this matters. When demographers say "cohort life expectancy," they mean: take everyone born in a particular year, follow them through their whole lives, and ask how long they lived on average. It's different from the number you usually see in headlines ("life expectancy at birth is 79"), which is a snapshot of current death rates applied to a hypothetical person. Cohort numbers are the truer, slower-cooked version. They tell you what actually happened — or, in a forecast, what's most likely to.
For a long time, each successive cohort in wealthy countries gained about 0.46 years of life expectancy over the one before it. Almost half a year, every cohort, like clockwork. The new paper says that pace is dropping by somewhere between 37% and 52%, depending on which forecasting method you trust. That's a wide range — and the authors are honest about it — but every method points the same direction.

Cohort life expectancy follows real generations through real lives — which makes any slowdown harder to wave away.
The surprising culprit: the very young
Okay, this is the part that genuinely surprised me. When you hear "life expectancy gains are slowing," you probably picture the same things I did — heart disease plateauing, obesity, the opioid crisis, all the grown-up problems. But the paper's age-decomposition analysis tells a different story. Over half of the total deceleration is attributable to mortality trends under age 5, and more than two-thirds is explained by mortality trends under age 20.
Read that again, because I had to. The slowdown isn't mostly about how 70-year-olds are doing. It's largely about the fact that the staggering 20th-century progress against infant and childhood mortality — vaccines, sanitation, neonatal care — was so successful that there's just less room left to improve. You can only drive a number so close to zero before the next cohort can't gain much by driving it closer. The authors note this pattern had already emerged in the observed data for the cohorts they analyzed, which is part of why they think it's unlikely to suddenly reverse.
The slowdown isn't mostly about how 70-year-olds are doing. It's about how spectacularly well we did against childhood mortality — and how little room that leaves for the next leap.
How worried should we actually be?
Honestly, the vibe of the paper is sober, not alarmist — and I want to match that here. A few things to hold in mind:
First, "deceleration" is not "decline." Life expectancy isn't predicted to fall. The gains are just expected to come more slowly. A generation still pulls ahead of the one before it — just by less.
Second, these are forecasts. Forecasts can be wrong. The researchers ran robustness checks and concluded the findings are unlikely to be solely due to downward bias in the modeling. But they're also careful: even if the numbers prove pessimistic, the underlying age pattern is already visible in real data. The direction of travel looks real, even if the exact speed is fuzzy.
Third — and this is the part I keep returning to — a slower societal escalator is exactly the case for taking the stairs.

If the population-level tailwind is fading, the everyday choices that compound across decades carry more weight, not less.
The reframing: personal choices, more leverage
Here's the thing nobody says out loud at longevity conferences: a lot of the spectacular gains of the last century were collective wins. Clean water. Childhood vaccines. Workplace safety laws. Antibiotics. You didn't earn those extra years; you were born into them. They showed up whether you went to the gym or not.
If the analysis is right, the easy collective gains are getting harder to come by. That doesn't mean the future is grim — it means the marginal year of healthy life is more likely to come from things that are within your reach: sleep, movement, what's on your plate, how you handle stress, whether you actually go to that screening appointment. The PNAS paper doesn't say any of this directly — it's a demography study, not a self-help manual. But the implication is hard to miss.
To be clear about what the evidence does and doesn't show: it's a moderate-strength finding. Multiple methods agree on the direction. The magnitude has a real range. And nothing in it speaks to any specific supplement, biohack, or wearable. Anyone selling you the slowdown as a reason to buy their thing is freelancing.
- The gains are slowing, not reversing. Multiple forecasts point to a 37–52% drop in the pace of cohort life expectancy improvement across 23 high-income countries.
- Childhood mortality is the unexpected driver. Over half the slowdown traces to mortality trends under age 5 — a sign of past success, not new failure.
- Forecasts are forecasts. The pattern is already visible in real data, but the exact size of the deceleration depends on the method.
- Collective wins are harder to come by now. Which makes individual habits — sleep, movement, food, screenings — relatively more important, not less.
- Talk to a clinician, not a TikTok. Personal longevity strategy is personal; it should involve someone who knows your actual health.
I started this piece a little gloomy and ended it weirdly motivated. The free ride your great-grandparents got from public-health miracles isn't going to keep giving each generation half a year for nothing. Fine. The flip side is that the things you can actually do — the boring, repeatable, deeply un-glamorous things — matter a little more than they did before. That's not a bad headline to live under.
Frequently asked questions
What is cohort life expectancy, and how is it different from the life expectancy figure I usually see reported?
Cohort life expectancy takes everyone born in a particular year, follows them through their whole lives, and measures how long they lived on average. The number typically seen in headlines is a snapshot of current death rates applied to a hypothetical person. The article describes cohort figures as 'the truer, slower-cooked version' because they reflect what actually happened to real generations.
How much has the pace of life expectancy gains actually slowed?
For a long time, each successive cohort in wealthy countries gained about 0.46 years of life expectancy over the one before it. The analysis found that pace is dropping by somewhere between 37% and 52%, depending on which forecasting method is used, across 23 high-income countries.
What is the main driver behind the slowdown in life expectancy gains?
Over half of the total deceleration is attributable to mortality trends under age 5, and more than two-thirds is explained by trends under age 20. The article explains this reflects the extraordinary success of 20th-century progress against infant and childhood mortality — vaccines, sanitation, and neonatal care — leaving very little room for further improvement in those age groups.
Does this mean life expectancy is expected to start falling?
No — the article is explicit that 'deceleration' is not 'decline.' Life expectancy is not predicted to fall; each generation is still expected to pull ahead of the one before it, just by less than previous generations did.
What does this slowdown mean for my own health habits?
The article argues that if the population-level tailwind is fading, individual habits carry more weight, not less. It points to sleep, movement, diet, stress management, and attending screening appointments as things within a person's reach, noting that the easy collective gains — clean water, vaccines, workplace safety laws — are getting harder to come by.
Sources
- Cohort mortality forecasts indicate signs of deceleration in life expectancy gains. — Proceedings of the National Academy of Sciences of the United States of America

Reversing the Decline: What the New Science Says About Aging Muscle
A 2025 review reframes sarcopenia as a modifiable condition — and grades which interventions actually move the needle on muscle mass, fiber loss, and motor unit decline.
For decades, the slow softening of the body after midlife was filed under the same heading as gray hair and reading glasses: inevitable. You lost a little muscle each year, your grip weakened, the stairs felt steeper, and the medical conversation, if it happened at all, was about accommodation rather than reversal. That framing is finally cracking. A 2025 review in the Journal of Functional Morphology and Kinesiology pulls together the documented trajectories of age-related muscle decline — and argues, with measured confidence, that much of what we call sarcopenia behaves less like an inevitability and more like a modifiable disease.
The shift matters most for women over 55, who tend to enter this decade with less baseline muscle than men and lose it against the headwind of menopausal hormonal change. The review's central claim is not that aging muscle can be made young again. It is subtler and, in some ways, more useful: the rate and depth of decline are not fixed, and several of the parameters clinicians track — muscle mass, Type II fiber size, motor unit firing rates — respond to the right interventions, in the right doses, even late in life.
What follows is a reader's guide to what the new synthesis actually says, where the evidence is strong, and where the honest answer is still we don't know yet.
- Decline is real but not linear. Muscle mass, Type II (fast-twitch) fibers, and motor unit firing rates each follow their own trajectory after midlife.
- Sarcopenia is being reclassified. The 2025 review treats it as a modifiable condition, not a fixed feature of aging.
- Strength training leads the evidence. Multimodal programs anchored by resistance work show the clearest signal for maintaining or improving function.
- Protein and progressive load matter together. Neither alone matches what they do in combination.
- The evidence is moderate, not definitive. Effect sizes vary by population, baseline, and program design.
- Start where you are. Benefits have been documented even in adults beginning training in their seventies and eighties.
What is actually declining
The review's most useful contribution is taxonomic. "Losing muscle" is shorthand for at least three distinct processes, and conflating them obscures what interventions can and cannot do.
The first is gross muscle mass — the cross-sectional acreage of tissue, measurable on imaging. The second is the preferential atrophy of Type II fibers, the fast-twitch units recruited for power: rising from a chair, catching yourself mid-stumble, lifting a grandchild. The third is motor unit decline — the nerves that fire those fibers drop out or fire less efficiently, so even the muscle that remains is incompletely activated. The review documents rates of decline across all three parameters, and they do not move in lockstep.
This is why a woman in her sixties can look unchanged in the mirror and still find that she cannot rise from the floor without a hand. Power — the product of force and speed — fades faster than mass. The fibers and motor units that produce it are the first to thin.

Grip and power decline earlier and faster than overall muscle mass — which is why function, not size, is the more telling measure.
Power fades faster than mass. The fibers that produce it are the first to thin — and the first to respond when you ask them to work again.
What the evidence says actually works
Here the review is careful, and we should be too. The strongest signal in the literature is for multimodal interventions anchored by strength training. The authors conclude that such programs can effectively maintain or improve physical function in aging adults, and in some studies appear to alter the trajectory of decline rather than merely slow it.
Three features tend to recur in the programs that show benefit. Loads are progressive — meaning resistance increases as the body adapts, rather than staying at a comfortable plateau. Training targets the Type II fibers specifically, which generally requires heavier loads or more explosive movement than the light weights long recommended to older women. And protocols are sustained; the gains are real but not permanent, and detraining undoes them on a timeline of weeks, not years.
Protein intake is the supporting actor that keeps showing up in the script. Older muscle is somewhat resistant to the anabolic signal that dietary protein provides, which is why the threshold to trigger muscle protein synthesis appears higher after midlife than before it. The review situates nutritional support — adequate protein, attention to vitamin D status — as a complement to mechanical loading, not a substitute for it. No supplement has been shown to replicate what lifting does.

Progressive resistance — not light, repetitive movement — is what consistently shows up in the trials that move the needle.
What the review does not claim
A moderate evidence rating means exactly that: real, replicated, but not yet definitive. The 2025 synthesis does not promise that any woman can fully reverse decades of decline, and it does not endorse a single optimized protocol that works for everyone. Effect sizes vary by starting point, by program design, by adherence, and by the specific outcome being measured. A program that adds measurable muscle mass may produce a more modest change in motor unit firing; a program that restores power may not visibly change the scale or the tape measure.
The review also stops short of prescribing. It synthesizes what the literature shows; it does not tell any individual reader what her loads, frequencies, or macronutrient targets should be. Those decisions belong in conversation with a clinician or a qualified trainer who knows your history — particularly if you are managing osteoporosis, cardiovascular disease, joint replacements, or the cluster of conditions that often accompany the years when this work matters most.
Why the reframing matters
For a generation of women who were told, often dismissively, that fatigue and weakness were simply what happens, the most important shift in this literature may be conceptual. Treating sarcopenia as a modifiable condition — rather than the price of being alive long enough — changes the questions worth asking. It changes what a useful annual physical might include. It changes what counts as a reasonable expectation for a seventy-year-old's next decade.
The science is not promising eternal youth. It is offering something more grounded and more honest: that the muscle you have at sixty-five is not the muscle you are stuck with at seventy-five, and that the interventions with the best evidence are the ones humans have always had access to — load, food, sleep, and the willingness to keep asking the body to do hard things. That is a moderate claim, made on moderate evidence. It is also, for a great many readers, the most useful sentence in the new literature.
The muscle you have at sixty-five is not the muscle you are stuck with at seventy-five.
Frequently asked questions
What does it actually mean to "lose muscle" as you age — isn't it all the same thing?
The article distinguishes at least three separate processes: the loss of overall muscle mass, the preferential shrinkage of Type II fast-twitch fibers responsible for power movements, and the decline of motor units — the nerves that fire those fibers, which drop out or fire less efficiently over time. These three processes do not move in lockstep, which is why overall muscle size can appear unchanged while functional ability falls noticeably.
Why might a woman in her sixties look the same in the mirror but suddenly find it hard to get up off the floor?
Because power — the product of force and speed — fades faster than gross muscle mass. The Type II fibers and motor units that produce explosive, quick movements are the first to thin, so functional ability declines before any visible change in size.
What do the exercise programs that show the clearest benefit have in common?
The review identifies three recurring features: loads are progressive, increasing as the body adapts rather than staying at a comfortable plateau; training specifically targets Type II fibers, which generally requires heavier loads or more explosive movement than light weights; and programs are sustained, because the gains reverse on a timeline of weeks once training stops.
Does eating more protein alone make up for not exercising?
No — the review frames nutritional support, including adequate protein and attention to vitamin D status, as a complement to mechanical loading, not a substitute for it. It also notes that older muscle is somewhat resistant to the anabolic signal protein provides, so the threshold needed to trigger muscle protein synthesis appears higher after midlife than before.
Does the review promise that older women can fully reverse decades of muscle loss?
No. The review carries a moderate evidence rating and explicitly does not promise full reversal or endorse a single protocol that works for everyone, noting that effect sizes vary by starting point, program design, adherence, and the specific outcome being measured. It also stops short of prescribing loads, frequencies, or nutritional targets for any individual.
Sources
- Reversing Decline in Aging Muscles: Expected Trends, Impacts and Remedies. — Journal of functional morphology and kinesiology

The Sarcopenia Signal: Why Your Nerves, Not Just Your Muscles, Drive Age-Related Decline
A new systematic review reframes age-related strength loss as a problem of failing wires, not shrinking cables — and it changes what screening and training after 60 should look like.
For most of the last century, the textbook story of aging muscle was a story about muscle. Fibers thinned, mitochondria sputtered, protein synthesis slowed, and somewhere around the seventh decade the body began the long negotiation we call sarcopenia. But a growing body of electromyography research is quietly rewriting that script. The first thing to fail, it turns out, may not be the muscle at all. It may be the wire that talks to it.
That, at least, is the through-line of a 2025 systematic review in GeroScience that pulled together 53 EMG studies on motor unit and neuromuscular junction (NMJ) function across aging and sarcopenia. The authors describe a recognizable sequence: early instability at the NMJ — the tiny synapse where a motor neuron hands off its electrical message to a muscle fiber — followed by the loss of motor units, then a period of compensatory remodeling in which surviving neurons attempt to adopt orphaned fibers. Overt atrophy and weakness, the clinical signs we currently use to diagnose sarcopenia, appear later in the cascade, not at its start. The implication is that by the time a grip-strength test catches the problem, the upstream pathology has been running for years.
The evidence here is moderate, not definitive. A systematic review is a synthesis, not a randomized trial, and EMG-derived parameters — jitter, jiggle, motor unit number estimation (MUNE) — remain specialized measurements more familiar to neurologists than to the geriatric clinics where sarcopenia is usually identified. Still, the convergence is hard to ignore: across dozens of studies, aging neuromuscular junctions show transmission instability and motor unit loss before muscle atrophy becomes clinically obvious.
The wires before the cables
To appreciate why this matters, it helps to picture the architecture. A single motor neuron in the spinal cord branches outward to innervate a cluster of muscle fibers; together they form a motor unit, the smallest functional element of voluntary movement. The NMJ is the junction at each fiber where acetylcholine crosses a microscopic gap and triggers contraction. If that junction becomes unreliable — if release is jittery, if receptors are sparser, if the structural folds that amplify the signal flatten — the muscle fiber is, in effect, partially deafferented. It is still there. It just isn't being called on properly.
The review describes two biomarkers that may track this process from a blood draw rather than a needle EMG: C-terminal agrin fragment, a byproduct of NMJ remodeling, and neurofilament light chain, a marker of axonal injury. Neither is ready for the corner lab, and neither has been validated as a screening tool in healthy aging populations. But both suggest a near-future in which sarcopenia risk could be assessed before a patient ever notices they are slower up the stairs.

Grip strength is a late signal. By the time it dips, the upstream neuromuscular cascade has been underway for years.
What this means for training
If the NMJ is the early failure point, then interventions that preserve neural drive should matter as much as those that build cross-sectional area. The review notes that resistance and endurance training, nutritional support, and electrostimulation-based approaches have each shown signals of attenuating NMJ decline, while physical inactivity and hormonal changes — menopause among them — appear to accelerate it. None of this overturns the existing consensus that lifting heavy things, regularly, is the single most evidence-backed intervention for sarcopenia. What it does is sharpen the rationale. Heavy, intent-driven contractions are not just hypertrophic stimuli; they are demands placed on the motor unit itself, the kind of demand that may help maintain the synapse.
The corollary is less flattering. Low-effort movement — the daily-step minimum, the gentle group class — is unlikely to be enough on its own. Programming that recruits high-threshold motor units, whether through external load or through ballistic and power-oriented work, is plausibly the part of training doing the neural work. We are not in a position to prescribe sets and reps from this evidence base, and any individual program should be designed with a clinician or qualified coach who understands a person's medical history. But the direction of travel is clear: train the nerve, not just the muscle.
By the time a grip-strength test catches it, the upstream pathology has been running for years.
Screening, and the intrinsic-capacity frame
The neuromuscular reframing also dovetails with how the WHO has been pushing healthy-aging assessment. Intrinsic capacity (IC) — a composite of locomotion, cognition, vitality, sensory function, and psychological state — is meant to catch decline earlier and more holistically than disease-by-disease screening. A 2025 cross-sectional study of 4,274 adults in the Queenstown cohort in Singapore found that 29.2% had at least one IC deficit, rising stepwise from 10.3% in adults aged 20–39 to 74.5% in those 80 and older, with locomotion the most commonly affected domain.
That study isn't an NMJ paper, but it is a useful sanity check on the population scale of the problem and on what already predicts it. Low handgrip strength was associated with 1.68-fold odds of an IC deficit, and frailty with nearly 11-fold odds — both consistent with a model in which neuromuscular integrity is doing quiet, foundational work behind the scenes of healthy aging. Grip strength is a crude proxy, but it is cheap, fast, and already in use; pairing it with a locomotion screen may be the most realistic near-term application of this science in primary care.

Power and intent-driven loading are increasingly seen as the part of training that taxes the motor unit itself.
- The nerve fails first. A 2025 systematic review of EMG studies suggests NMJ instability and motor unit loss precede the muscle atrophy clinicians currently use to diagnose sarcopenia.
- The evidence is moderate. Findings come from a synthesis of 53 studies using specialized EMG measures; they are convergent but not yet trial-grade.
- Biomarkers are emerging. C-terminal agrin fragment and neurofilament light chain may eventually allow earlier risk detection, but neither is ready for routine use.
- Training should target neural drive. Resistance and endurance work, and possibly power-oriented loading, are the interventions with the strongest signal for preserving NMJ function.
- Screening is catching up. WHO intrinsic-capacity frameworks and handgrip testing already capture much of the downstream signal — and locomotion deficits are the most common in adults over 60.
- This is not medical advice. Anyone considering a meaningful change to training, supplementation, or screening should do so with a clinician who knows their history.
The big story here is not that we have a new treatment for sarcopenia. We don't. It is that the unit of analysis is shifting. For a long time, age-related decline has been framed as a slow leak from a muscle bucket, and the response has been to pour more in — more protein, more reps, more time under tension. The neuromuscular reframing suggests a different metaphor. The bucket isn't leaking. The pump is wearing out. That is a harder problem, but it is a more honest one, and it points toward earlier action, better screening, and training designed for the nervous system that drives the muscle, not just the muscle itself.
Frequently asked questions
Why is grip strength considered a late indicator of sarcopenia according to this research?
The article explains that NMJ instability and motor unit loss occur before muscle atrophy becomes clinically obvious, meaning the upstream neuromuscular cascade has been underway for years by the time a grip-strength test detects a problem. Grip strength reflects overt weakness, which appears later in the cascade rather than at its start.
What are the two blood biomarkers mentioned, and can they be tested today?
The article identifies C-terminal agrin fragment, a byproduct of NMJ remodeling, and neurofilament light chain, a marker of axonal injury, as emerging biomarkers. Neither is ready for routine clinical use, and neither has been validated as a screening tool in healthy aging populations.
What does the article say about low-intensity movement like daily steps or gentle group classes?
The article states that low-effort movement is unlikely to be enough on its own to address neuromuscular decline. Programming that recruits high-threshold motor units, through external load or ballistic and power-oriented work, is described as the part of training doing the neural work.
What factors does the article say can accelerate NMJ decline?
The article specifically names physical inactivity and hormonal changes, including menopause, as factors that appear to accelerate neuromuscular junction decline.
How large was the systematic review described in the article, and what did it find?
The 2025 systematic review published in GeroScience synthesized 53 EMG studies on motor unit and neuromuscular junction function across aging and sarcopenia. It describes a recognizable sequence of early NMJ instability, followed by motor unit loss, then compensatory remodeling by surviving neurons, with overt atrophy and weakness appearing later in the cascade.
Sources
- Exploring motor unit and neuromuscular junction dysfunction in aging and sarcopenia: insights from electromyography in systematic review. — GeroScience
- Intrinsic capacity deficits across the lifespan in a nationally representative community cohort: findings from the Queenstown study. — Archives of gerontology and geriatrics
Why Your Flu Shot Works Less Well at 70: The New Science of Fixing Vaccines for Older Immune Systems
Aging immune systems mount weaker responses to vaccines. A new Nature Aging review maps the engineering — higher doses, smarter adjuvants, mRNA — designed to close the gap.
Roll up a sleeve at 35 and your immune system tends to read the vaccine like a sharp memo: short, urgent, acted on. Roll up the same sleeve at 75 and the memo still arrives — but it lands on a desk piled with older mail, in an office where the staff has thinned out and the lights flicker. The shot is the same. The reader is not. That mismatch, increasingly called immunosenescence, is why the people who most need protection from influenza, RSV, and COVID-19 often get the least from the standard versions of the vaccines designed to provide it. A 2025 review in Nature Aging argues the fix is no longer hypothetical: it's a toolkit of higher-dose antigens, next-generation adjuvants, mRNA platforms, and — more speculatively — drugs that target aging itself.
- The problem is the host, not the pathogen. Aging immune cells respond less vigorously to the same vaccine antigen, a phenomenon called immunosenescence.
- More antigen helps, but isn't the whole answer. High-dose formulations are one of several strategies reviewed for boosting older-adult responses.
- Adjuvants and mRNA platforms are being engineered specifically for older immune systems, alongside efforts toward universal vaccines.
- Geroscience is the wild card. Early clinical signals suggest interventions like mTOR inhibition may improve vaccine responses — promising, but not yet standard care.
- Evidence rating: Moderate. The mechanisms are well-described and several strategies are clinically deployed; head-to-head long-term outcomes data is still maturing.
What actually goes wrong with an older immune response
The shorthand "immunosenescence" covers a lot of ground. It includes thymic involution (the organ that schools new T cells shrinks with age), a narrower repertoire of naive lymphocytes available to recognize novel threats, and a chronic low-grade inflammatory tone — sometimes called "inflammaging" — that paradoxically blunts the sharp, targeted response a vaccine is trying to provoke. The Nature Aging review frames these as overlapping problems: an older immune system isn't simply weaker, it's miscalibrated, with the baseline noise turned up and the signal-detection turned down. That is the substrate every vaccine in a 70-year-old arm has to work against, and it's why the same dose that protects a 30-year-old can underperform in a 70-year-old by a meaningful margin.
The clinical consequence is familiar to anyone who has watched a flu season tear through a long-term care facility despite high vaccination rates. The vaccine wasn't a fraud. The immune system reading it had aged.
Higher-antigen formulations are one of several strategies aimed at older immune systems.
Strategy one: more antigen
The most intuitive fix is also the bluntest: give the older immune system more of what it's supposed to recognize. High-dose influenza vaccines — already in use in many countries for adults 65 and over — embody this approach. The Nature Aging review treats increased antigen quantity as one pillar of a multi-pillar strategy, useful but not sufficient on its own. More antigen can partially compensate for a less responsive system, but it does not reverse the underlying cellular aging that limits how robust and durable the response becomes in older adults.
An older immune system isn't simply weaker. It's miscalibrated — baseline noise turned up, signal detection turned down.
Strategy two: smarter adjuvants
Adjuvants are the supporting cast — molecules added to a vaccine to recruit and energize the immune cells that will then respond to the antigen. The newer generation reviewed by the authors aims not just to amplify the response generally, but to engage pathways that age-related changes have left underused. The thinking is mechanistic: if the problem is that older immune cells are slower to mobilize and quicker to default to inflammation rather than precise targeting, an adjuvant chosen to coax precise targeting may matter more than one that simply turns the volume up across the board.

Strategy three: mRNA, and the universal-vaccine ambition
mRNA platforms — vaulted into household familiarity by the COVID-19 pandemic — are being investigated not only as a faster way to make vaccines but as a substrate that may engage older immune systems differently than traditional protein-based shots. The review groups mRNA with another long-running ambition: universal vaccines, designed to elicit immunity against the conserved parts of viruses (the regions that don't change much from strain to strain), potentially freeing older adults from the annual guessing game of matching that year's circulating influenza to that year's formulation. Both are characterized in the review as promising rather than proven for the older-adult use case specifically; the underlying biology is plausible and early data is encouraging, but the long-term, head-to-head clinical record in older populations is still being built out.
Strategy four: treat the aging, not just the vaccine
The most speculative — and most interesting — section of the review concerns geroscience: the idea that interventions targeting aging biology itself could make whatever vaccine you give work better. The authors discuss emerging clinical evidence around mTOR inhibition and caloric restriction as candidate approaches, on the logic that they may attenuate chronic inflammation and the metabolic shifts that accompany immune aging in older adults. This is genuinely early science. It is not a regimen to adopt; it is a research direction to watch. The signal in the review is that the field is increasingly willing to ask whether the right intervention point is the vaccine, the immune system, or the aging process feeding both.
How strong is the evidence, really?
Moderate, and worth being precise about why. The biology of immunosenescence is well-described and reproducible. Several of the engineering strategies — higher-dose formulations, certain adjuvanted vaccines — are already in clinical use in older adults, supported by trials showing improved immunogenicity and, in some cases, improved clinical outcomes versus standard formulations. mRNA and universal-vaccine approaches are advancing but the older-adult-specific long-term efficacy data is still being generated. The geroscience strategies are the most preliminary: mechanistically compelling, with early clinical signals, but a long way from standard practice. The Nature Aging review's contribution is to set these in one frame — not to declare the problem solved, but to show that the toolkit for solving it is no longer empty .
The honest summary: the next decade of vaccine development for older adults will likely look less like one breakthrough and more like a layered set of fixes — more antigen here, a better adjuvant there, a new platform somewhere else, and, eventually perhaps, a pill that makes the immune system a little less old before the needle goes in.
Frequently asked questions
Why do standard vaccines work less well in older adults?
As the immune system ages, a process called immunosenescence leaves it miscalibrated rather than simply weaker — the baseline inflammatory noise is turned up while the ability to mount a sharp, targeted response is turned down. This means the same vaccine dose that protects a 30-year-old can meaningfully underperform in a 70-year-old.
What is 'inflammaging' and how does it affect vaccine response?
Inflammaging refers to the chronic low-grade inflammatory tone that develops with age. According to the article, this paradoxically blunts the precise, targeted immune response a vaccine is trying to provoke, making it harder for older immune systems to respond effectively to vaccination.
Do higher-dose flu vaccines fully solve the problem for older adults?
High-dose influenza vaccines can partially compensate for a less responsive immune system, but the article describes increased antigen quantity as one pillar of a multi-pillar strategy — useful but not sufficient on its own. More antigen does not reverse the underlying cellular aging that limits how robust and durable the response becomes.
What role might mRNA vaccines play in protecting older adults?
mRNA platforms are being investigated as a substrate that may engage older immune systems differently than traditional protein-based shots. The article characterizes this as promising rather than proven for older adults specifically, noting that long-term, head-to-head clinical data in older populations is still being built out.
Are the geroscience strategies discussed in the article something older adults should try?
No. The article explicitly states that approaches like mTOR inhibition and caloric restriction are not approved or recommended for boosting vaccine response — they are a research direction to watch, not a regimen to adopt. Anyone over 65 with questions about appropriate vaccine formulations should consult a physician or pharmacist.
Sources

Menopause Timing May Be an Early Warning Light for Brain Aging
A large multiomic study links later menopause with healthier prefrontal cortex aging decades on — a finding worth knowing for the women in your life, and for what it suggests about how the body ages as one system.
The body, it turns out, keeps its books across organs. A finding from Aging Cell suggests that something as seemingly distant as the timing of a woman's last menstrual period may carry information about how her prefrontal cortex — the part of the brain that plans, judges and remembers — will age decades later. For those of us north of sixty, this is not abstract. It concerns wives, sisters, daughters, and, frankly, the way we ought to think about aging itself: not as a sequence of unrelated failures, but as one slow, connected process we can sometimes read in advance.
The study in question is a multiomic analysis of 2,086 post-mortem brain samples, asking a simple question with complicated machinery behind it: does age at menopause (AAM) — a marker of ovarian aging — predict how the brain ages later on? The short answer from the researchers is yes, modestly and meaningfully. Later menopause was positively correlated with cognitive function and negatively correlated with prefrontal cortex aging acceleration, the latter measured as biological age from DNA methylation minus chronological age. In plain English: women whose ovaries kept working longer tended to have brains that looked, molecularly, a little younger than their birthdays.
That is the headline. The fine print is what makes it interesting.
What the omics actually showed
The authors went looking for mechanism, not just association. Transcriptomic data — the readout of which genes are switched on — confirmed that later age at menopause was associated with prefrontal cortex gene expression consistent with better cognition. Among women who reached menopause naturally, later timing also tracked with reduced expression of pathways implicated in aging itself.
Women whose menopause was surgical — typically through removal of the ovaries — looked different at the molecular level. The researchers flagged perturbed nicotinamide adenine dinucleotide (NAD+) activity, validated by metabolomics, and disturbances in bile acid metabolism in both natural and surgical groups, though with different bile acid ratios involved. NAD+ is a coenzyme central to how cells produce energy and repair DNA; bile acids, beyond digestion, increasingly look like signaling molecules with reach into the brain. None of this proves causation. It does suggest the conversation between ovaries and brain runs through pathways we are starting to map.

Multiomic studies stitch together DNA methylation, gene expression and metabolites — the closest thing we have to a wide-angle lens on how the body ages.
Why this matters even if you are not the patient
Dementia risk prediction beyond age itself is, as the authors put it bluntly, challenging. A new case is diagnosed roughly every three seconds globally. Most of the tools we have — cognitive testing, imaging, spinal fluid markers — catch trouble fairly late. The promise of an ovarian-aging signal is that it shows up decades earlier, in a piece of information a woman already knows about herself. If it holds up in living cohorts, it would not be a diagnosis. It would be a nudge — a reason to take blood pressure, sleep, hearing, exercise and the other modifiable risks more seriously, sooner.
For the men in this audience, there are two practical implications. The first is conversational: if your spouse or sister had an early natural menopause or a surgical one, that is worth her mentioning to her own clinician as part of a long-view brain-health discussion. The second is conceptual. Cross-organ aging signals — the idea that one tissue's clock tells you something about another's — are likely coming for us too. Erectile function, grip strength, gait speed, hearing thresholds: each is being studied as a window onto systemic aging. The menopause finding is a particularly clean version of a pattern we should expect to see more of.
The body keeps its books across organs. The trick is learning to read them early.
What this study is — and is not
A measured reading is in order. This is post-mortem tissue, not a clinical trial. The authors lean on genetic correlations suggesting shared heritability between menopause timing and brain aging, which strengthens the case that we are looking at biology rather than coincidence, but shared genes are not the same as a proven mechanism. Correlations were present and consistent; effect sizes in this kind of work are typically modest. Nothing here says that a woman whose periods stopped at 48 is destined for cognitive decline, or that one who finished at 55 is protected. It says the curves, on average, tilt.
Nor does the study tell us what to do about it. The hormonal therapy question — whether estrogen replacement after early or surgical menopause influences this trajectory — is not answered here and remains a decision for a woman and her clinician, weighing her own history. The NAD+ signal will inevitably be seized on by the supplement industry; the study does not show that taking NAD+ precursors changes brain aging in humans, and we should be careful not to let one molecular clue become tomorrow's marketing campaign.

Family history of menopause timing is a piece of information most women already carry — and one worth bringing to a primary-care visit.
- The finding. In 2,086 post-mortem brains, later age at menopause tracked with a younger-looking prefrontal cortex and better cognitive markers.
- The mechanism, in part. Gene expression, NAD+ activity and bile acid metabolism all showed differences tied to menopause timing — especially in women with surgical menopause.
- The strength of evidence. Moderate. Strong study size and multiomic depth, but observational and based on tissue at death rather than living cohorts.
- What it is not. Not a diagnosis, not a prescription, not a green light for NAD+ supplements.
- What to do. Women — especially those with early or surgical menopause — can raise menopause timing with their clinician as one input into a long-view brain-health plan.
- The bigger idea. Aging in one organ often whispers about aging in another. Expect more findings like this — for both sexes.
The deeper takeaway from this kind of research is the one easiest to miss in the headlines: the body ages as a system, and the system leaves clues. We are not going to outrun aging. But the gap between knowing a risk at 55 and knowing it at 75 is, in practice, the difference between adjusting and reacting. That gap is where longevity medicine actually lives, and it is where studies like this one — careful, mechanistic, honest about their limits — quietly earn their keep.
Frequently asked questions
What did the study actually find about menopause timing and brain aging?
In 2,086 post-mortem brain samples, later age at menopause was positively correlated with cognitive function and negatively correlated with prefrontal cortex aging acceleration, measured as biological age from DNA methylation minus chronological age. In plain terms, women whose ovaries kept working longer tended to have brains that looked, molecularly, a little younger than their birthdays.
Does an early menopause mean a woman is destined for cognitive decline?
No. The article is explicit that nothing in the study says a woman whose periods stopped at 48 is destined for cognitive decline, or that one who finished at 55 is protected. The finding means the curves, on average, tilt — effect sizes in this kind of work are typically modest, and the study is observational rather than a clinical trial.
Was surgical menopause different from natural menopause in this research?
Yes. Women whose menopause was surgical looked different at the molecular level, with researchers flagging perturbed NAD+ activity validated by metabolomics and disturbances in bile acid metabolism. Both natural and surgical groups showed bile acid differences, though with different bile acid ratios involved.
Should women start taking NAD+ supplements based on this finding?
The article cautions against it, noting that the study does not show that taking NAD+ precursors changes brain aging in humans. It warns that one molecular clue should not become tomorrow's marketing campaign, and explicitly states that NAD+ supplements are not a green light from this research.
What is the practical takeaway for a woman reading this?
According to the article, women — especially those with early or surgical menopause — can raise menopause timing with their clinician as one input into a long-view brain-health plan. The article also notes that family history of menopause timing is a piece of information most women already carry and one worth bringing to a primary-care visit.
Sources

Inheriting Resilience: Could a Parent's Mitochondrial Stress Make You Live Longer?
A new worm study suggests that mild stress in one generation can leave longevity marks on the next. The mechanism is fascinating — and the leap to humans is still a long one.
For more than a century, biologists have flirted with a heretical idea: that the experiences of one generation might leave a fingerprint on the next, written not in DNA letters but in the molecular scaffolding that decides which letters get read. A new paper in Redox Biology pushes that idea into sharper focus. Working with the millimeter-long roundworm Caenorhabditis elegans, researchers report that a carefully measured dose of mitochondrial stress in parent worms not only extends the parents' own lifespan but also hands a longevity advantage down to offspring who were never exposed — and they trace the inheritance to specific chemical tags on histone proteins. It is a striking finding. It is also, importantly, a finding in worms.
- What the study shows: In C. elegans, mild mitochondrial oxidative stress in parents extended lifespan in both the parents and their unexposed progeny.
- How it travels: The effect depended on the mitochondrial unfolded-protein response (UPRmt) and two stress-response transcription factors, DAF-16/FOXO and SKN-1/Nrf2.
- The epigenetic carriers: Two histone marks — H3K4me3 (activating) and H3K27me3 (repressing) — selectively tuned genes tied to oxidative-stress response and longevity.
- What it does not show: Anything about humans. The work is preclinical and conducted entirely in a short-lived invertebrate.
- Why it still matters: It sharpens a mechanistic case for hormesis — the idea that small, well-dosed stressors can build resilience.
Hormesis, in one paragraph
Hormesis is the principle that a stressor lethal at high doses can be beneficial at low ones. Exercise damages muscle fibers; the repair makes you stronger. Brief heat or cold exposure provokes a stress response that, over time, appears to recalibrate cellular housekeeping. Mitochondria — the organelles that turn food and oxygen into usable energy — are central to this story. When they're nudged just hard enough to leak a little reactive oxygen, cells often respond by upgrading their defenses rather than breaking down. The new Redox Biology paper asks a question that hormesis researchers have circled for years: does that upgrade stay with the individual, or can it travel?
What the worms actually did
The team, led by Wan and colleagues, exposed parent worms to mitochondrial hormetic oxidative stress (which they abbreviate mtHOS) and tracked both the exposed animals and their descendants. The exposed parents lived longer, as prior hormesis work would predict. The novel observation was that their progeny — never themselves exposed — also lived longer, a pattern the authors describe as transgenerational epigenetic inheritance of the longevity signal.
Mechanistically, the inheritance depended on a coordinated cast of molecular players. The mitochondrial unfolded-protein response (UPRmt), a quality-control program that kicks in when mitochondria are under strain, had to be active. Two transcription factors — DAF-16, the worm version of the FOXO family, and SKN-1, the worm counterpart of mammalian Nrf2 — had to be working in concert. And at the chromatin level, two opposing histone marks did the bookkeeping: H3K4me3, which generally flags genes for active transcription, and H3K27me3, which generally damps genes down. Together, the authors report, these marks selectively regulated genes tied to oxidative-stress response and longevity, effectively pre-tuning the next generation's stress vocabulary before it had encountered any stress of its own.

Histones — the spool-like proteins that package DNA — carry chemical marks that tell cells which genes to read loudly and which to whisper.
The worms inherited a tuned stress response without inheriting the stress itself. On the central finding
Why this is more than a curiosity
Transgenerational epigenetic inheritance is well documented in C. elegans and has been demonstrated, less tidily, in some plant and rodent systems. Its existence in mammals — and certainly in humans — remains contested terrain. Worms have biological features that make the inheritance comparatively clean to study: a short life cycle, a transparent body, hermaphroditic reproduction, and a germline that interacts with somatic stress signals more readily than in mammals. None of those features carry over to people.
That caveat aside, the worm result is mechanistically interesting because it identifies which marks matter and which upstream programs the marks depend on. The authors point to a coordinated axis of UPRmt activation plus DAF-16/FOXO and SKN-1/Nrf2 signaling, with H3K4me3 and H3K27me3 serving as the durable record. Those pathways have mammalian counterparts that are already targets of intense longevity research. The worm paper is not evidence that the same inheritance happens in humans; it is evidence that, in at least one organism, the inheritance has an identifiable molecular address.
What it does — and does not — say about exercise, heat and cold
The framing temptation here is obvious. If mild mitochondrial stress in parents can hand resilience to offspring, what about the mild mitochondrial stressors many readers are already curious about — endurance exercise, sauna sessions, cold plunges? The honest answer is that this paper does not test any of those interventions, in any species, in any generation. It tests a defined laboratory stressor in worms.
What the paper does is strengthen the mechanistic plausibility of hormesis as a category. It suggests that the cellular machinery worms use to bank a stress response — UPRmt, FOXO-family and Nrf2-family signaling, histone methylation — is doing something coherent and durable, not just twitching in response to a transient insult. For readers already weighing GLP-1 medications, structured exercise, or thermal exposure as parts of a long-term metabolic strategy, the takeaway is modest but real: the biology of small, repeated stressors is becoming better mapped, and that map is starting to include how the marks persist.

Exercise is the best-studied human hormetic stressor. The worm work does not test it — but it makes the underlying logic harder to wave away.
What would have to be true for this to matter to humans
Several layers of evidence are missing between this worm result and any human implication. First, researchers would need to show that comparable hormetic stressors in mammals produce comparable histone-mark patterns in the germline. Second, they would need to show that those marks survive the extensive epigenetic reprogramming that occurs in mammalian embryos — a process that erases much of what is written. Third, they would need outcome data: not just marks, but measurable differences in healthspan or disease risk in offspring. None of that work is done.
It is also worth being precise about what the worm paper claims and what it does not. The authors describe a sophisticated interplay among oxidative-stress response genes and chromatin remodeling that enhances progeny resilience to future challenges. They do not claim a universal mechanism, a human translation, or a dosing schema. Readers should resist the temptation to fill in those blanks.
Mechanism in worms is a starting line, not a finish line.
A better conversation with your clinician
If this study changes anything for the average reader, it is the texture of the conversation rather than the prescription. Hormesis-adjacent practices — structured exercise, supervised heat or cold exposure, dietary patterns that include periods of mild metabolic stress — already have human evidence behind them at varying strengths. The new paper does not upgrade any of those into proven longevity interventions. It does add another mechanistic reason to take the category seriously, and another reason to be skeptical of products that promise the benefits of stress without any of the stress.
For anyone on or considering a GLP-1 medication, the relevance is indirect but real. GLP-1 therapy changes the body's energy economy, and the lifestyle scaffolding around it — particularly resistance training and aerobic exercise — is where most of the durable metabolic benefit is likely to come from. Mechanistic work like this paper is a reminder that those stressors are doing biochemical work worth respecting, even when their payoff is slow.
- The finding is real but narrow: a worm study, in a well-defined experimental setup, with a clear molecular mechanism.
- The translation to humans is unproven. Mammalian germline reprogramming is a serious barrier the paper does not address.
- Hormesis is a category, not a prescription. Talk to a clinician before adding heat, cold or aggressive exercise protocols, especially on GLP-1s.
Frequently asked questions
What did the study actually find?
Working with the roundworm C. elegans, researchers found that a carefully measured dose of mitochondrial stress in parent worms extended lifespan in both the exposed parents and their offspring who were never themselves exposed to that stress. The team traced this inherited longevity advantage to specific chemical tags on histone proteins, identifying a coordinated pathway involving the mitochondrial unfolded-protein response and two stress-response transcription factors.
What is hormesis, and why does it matter here?
Hormesis is the principle that a stressor lethal at high doses can be beneficial at low ones — for example, exercise damages muscle fibers, but the repair process makes you stronger. The study strengthens the mechanistic case for hormesis by suggesting that the cellular machinery worms use to bank a stress response is doing something coherent and durable, not just reacting to a transient insult.
How exactly does the longevity signal get passed from parent worms to their offspring?
The inheritance required the mitochondrial unfolded-protein response (UPRmt) to be active, along with two transcription factors — DAF-16/FOXO and SKN-1/Nrf2 — working in concert. At the chromatin level, two opposing histone marks — H3K4me3, which flags genes for active transcription, and H3K27me3, which damps genes down — selectively regulated genes tied to oxidative-stress response and longevity, effectively pre-tuning the next generation's stress response before it encountered any stress of its own.
Does this study tell us anything about what humans should do?
No — the work is preclinical and conducted entirely in a short-lived invertebrate, and the article explicitly states it does not show anything about humans. Animal-preclinical evidence like this can identify mechanisms and generate hypotheses, but cannot on its own justify a clinical recommendation.
What would need to happen before this worm finding could have implications for people?
Researchers would first need to show that comparable hormetic stressors in mammals produce comparable histone-mark patterns in the germline. They would then need to show that those marks survive the extensive epigenetic reprogramming that occurs in mammalian embryos — a process the article notes erases much inherited molecular information.
Sources

PRP, Stem Cells, and Peptides: Where Regenerative Orthopedics Actually Has Evidence
A 2025 review of 160 studies grades four regenerative modalities for joint and tendon care. The signal is real — but uneven, and narrower than the marketing suggests.
Walk past any high-end orthopedic clinic in 2026 and you will see the same vocabulary on the window: platelet-rich plasma, stem cells, peptides, biomimetic scaffolds. The promise is seductive — repair instead of replace, regenerate instead of resect. The marketplace has expanded faster than the evidence underneath it, which leaves the curious reader with a genuinely hard question: which of these therapies has actually earned its place in a serious recovery plan, and which is selling a centrifuge dressed up as a cure?
A structured review published this year in the Journal of Clinical Medicine offers the most useful map yet. The authors screened roughly 160 studies from PubMed between 2009 and January 2025, then narrowed to 59 that met inclusion criteria prioritizing randomized controlled trials — 20 on platelet-rich plasma (PRP), 20 on mesenchymal stem cells (MSCs), 10 on peptide therapies, and 7 on biomimetic materials. Risk of bias was assessed with the Cochrane and ROBINS-I tools, and a random-effects meta-regression weighed therapy type, sample size, and bias against reported pain outcomes. It is, in other words, the kind of synthesis a longevity-minded reader should actually read before booking an injection.
The headline finding: not all four modalities are created equal, and the gap is wider than clinic brochures imply. In the meta-regression, MSC therapy emerged as the most effective intervention for pain reduction (β = 8.45, p < 0.05). PRP and peptide-based therapies showed moderate improvements. Biomimetic materials — the engineered scaffolds meant to coax tissue back into shape — landed at the bottom of the effect-size ranking. None of this means biomimetics are a dead end; it means the human clinical evidence is still thin and early.
PRP: a short-acting tool, not a cure
Platelet-rich plasma is the most familiar name on the list because it is the easiest to deliver: spin a patient's blood, concentrate the platelets, inject. The review's read on PRP is measured rather than dismissive. It provided short-term pain relief, particularly in acute injuries and tendon repair. That is a real, useful finding — and also a narrower claim than what is often advertised. The same review flagged that inconsistencies in preparation methods limited success in chronic conditions, which is the polite scientific way of saying that two clinics calling something "PRP" may be delivering meaningfully different products.
For the reader trying to translate this into a decision, the practical takeaway is unglamorous: PRP appears most defensible as a short-horizon intervention for an acute tendon or soft-tissue problem, and least defensible as a recurring maintenance ritual for chronic joint pain. Preparation protocol, platelet concentration, and leukocyte content are not trivial details; they are the intervention.
PRP's strongest signal is in acute tendon and soft-tissue injury — not chronic joint maintenance.
MSCs: the strongest signal, with the biggest caveats
Mesenchymal stem cells are where the review's enthusiasm and its disclaimers coexist most uncomfortably. The pain-reduction effect was the largest of the four modalities, which is exactly the result the regenerative field has been chasing for two decades. But effect size in a meta-regression is not the same as a finished clinical playbook. Cell source (bone marrow, adipose, umbilical), dose, expansion technique, and delivery route still vary widely across the studies that fed into the analysis, and the regulatory status of many MSC products remains unsettled.
What this means for a reader evaluating a clinic: the underlying biological signal is real and increasingly hard to wave away, and the operational details — where the cells came from, how they were processed, how they were delivered — determine whether you are buying into that signal or into something only loosely related to it. The right posture is curiosity with a high bar for documentation, not faith.
The biology is real. The product you are buying may or may not be. Felix Mercer
Peptides and biomimetics: promising, premature
Peptide-based therapies — short amino-acid sequences designed to nudge specific repair pathways — landed in the moderate-improvement bucket alongside PRP. With only 10 trials meeting inclusion criteria, the evidence base is genuinely small, and the marketing is genuinely loud. The mismatch matters. A modest, replicated signal in a handful of RCTs is a reason to follow the field; it is not a reason to assemble a multi-peptide stack on the basis of a podcast.
Biomimetic materials sit further upstream still. Only seven studies cleared the review's bar, and the modality showed the lowest effect in the pain-reduction analysis. That is not a verdict on the underlying science — engineered scaffolds for cartilage and bone are an active and serious area of research — but it is a verdict on what the human clinical record currently supports. Today, biomimetics belong in trials, not on a consumer menu.
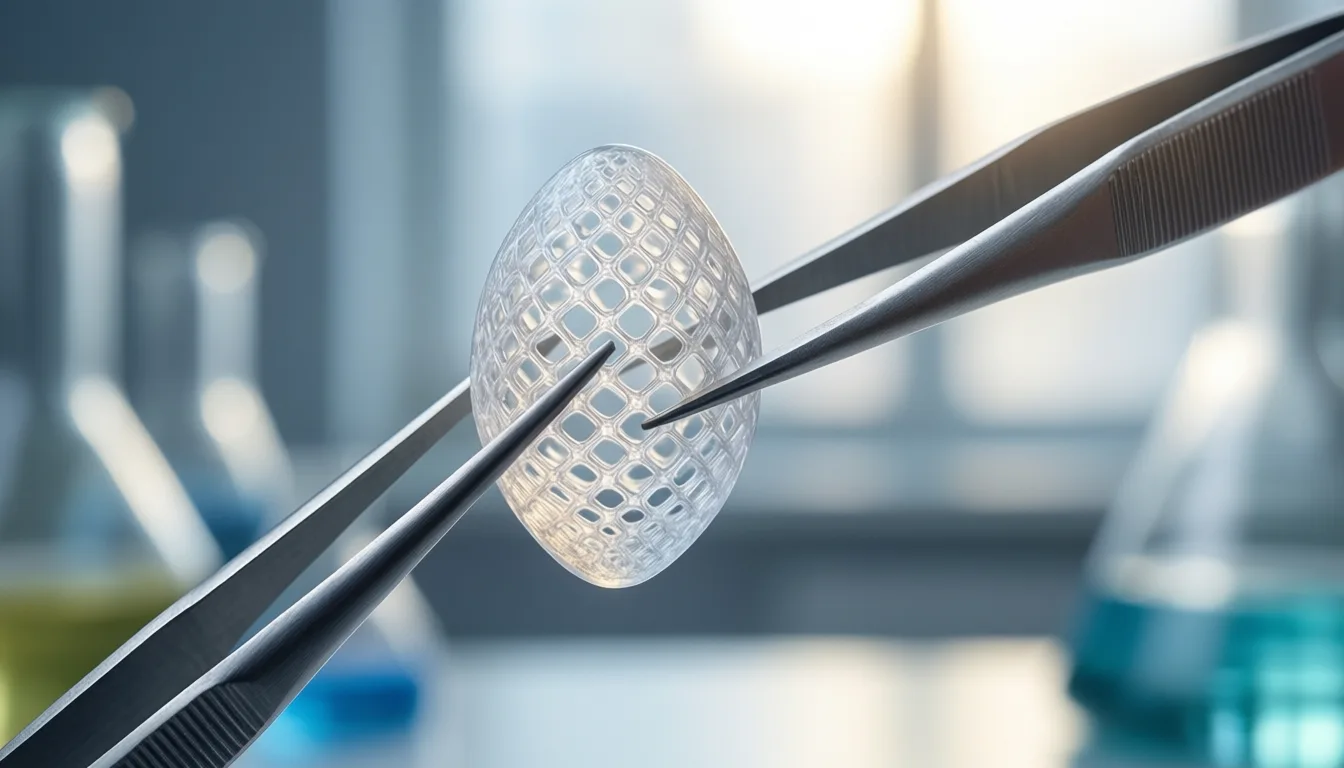
Biomimetic scaffolds are scientifically serious and clinically early — a field to watch, not to buy.
How to read a regenerative clinic
The most useful thing a 160-study review can give a reader is not a verdict but a vocabulary. If a clinic cannot tell you which preparation protocol they use, which cell source, which peptide sequence, which scaffold material — and what the comparator was in the trials they cite — you are not being offered evidence-based care. You are being offered a procedure that resembles one. The four modalities here are not equivalent, the trials behind them are not equivalent, and the clinic delivering them is not incidental to the outcome.
None of this is medical advice, and none of it substitutes for a conversation with a clinician who knows your imaging, your history, and your goals. But it is a defensible frame for that conversation: ask which modality the evidence actually supports for your specific problem, ask what the protocol is, and ask what the alternative — including conservative care — would look like over the same horizon.
- MSCs led the 2025 meta-regression for pain reduction (β = 8.45, p < 0.05), but product variability remains the central caveat.
- PRP is best read as short-term, with the clearest signal in acute injury and tendon repair, not chronic joint maintenance.
- Peptide therapies show moderate effects across a small evidence base (10 trials) — a field to follow, not a stack to assemble.
- Biomimetic materials are early: only 7 included trials and the lowest measured effect on pain.
- Preparation and protocol are the intervention. Two clinics offering the "same" therapy may be delivering meaningfully different products.
- This is education, not prescription. Bring the framing — and the questions — to a clinician who knows your case.
Frequently asked questions
Which of the four regenerative therapies showed the strongest evidence for pain reduction?
Mesenchymal stem cell (MSC) therapy emerged as the most effective intervention for pain reduction in the meta-regression, with an effect size of β = 8.45 (p < 0.05). PRP and peptide-based therapies showed moderate improvements, while biomimetic materials landed at the bottom of the effect-size ranking.
Is PRP a good option for chronic joint pain?
According to the review, PRP appears most defensible as a short-horizon intervention for acute tendon or soft-tissue problems and least defensible as a recurring maintenance ritual for chronic joint pain. Inconsistencies in preparation methods also limited its success in chronic conditions.
Why does it matter which clinic delivers a regenerative therapy?
Preparation protocol, platelet concentration, and leukocyte content are not trivial details — they are the intervention itself. Two clinics offering the same therapy may be delivering meaningfully different products, and operational details like cell source, processing method, and delivery route affect whether a patient is buying into the evidenced biological signal or something only loosely related to it.
How strong is the evidence behind peptide therapies?
Peptide-based therapies showed moderate improvement in the analysis, but only 10 trials met the review's inclusion criteria, making the evidence base genuinely small. The article notes this is a reason to follow the field, not to assemble a multi-peptide stack based on a podcast.
Why are biomimetic materials not yet considered a consumer option?
Only seven biomimetic studies cleared the review's inclusion bar, and the modality showed the lowest effect in the pain-reduction analysis. The article describes them as scientifically serious but clinically early — a field to watch, not to buy.