
In This Issue
Metabolic Health
-
 Cardiometabolic Resilience: Who Avoids the Damage, and Why It Matters for You
Two new datasets reframe metabolic risk from inevitability to variability — hunting the protective phenotypes that shrug off vascular damage, and the routine labs that flag your five-year diabetes window.
Cardiometabolic Resilience: Who Avoids the Damage, and Why It Matters for You
Two new datasets reframe metabolic risk from inevitability to variability — hunting the protective phenotypes that shrug off vascular damage, and the routine labs that flag your five-year diabetes window.
-
 Rewiring Glucose Control: What Three New Papers Say About Gut, Liver, and Diabetes
Microbiota-targeted therapies, a newly mapped liver enzyme called HYAL1, and Mendelian evidence linking metabolic syndrome to IBD are quietly reshaping how scientists think about type 2 diabetes.
Rewiring Glucose Control: What Three New Papers Say About Gut, Liver, and Diabetes
Microbiota-targeted therapies, a newly mapped liver enzyme called HYAL1, and Mendelian evidence linking metabolic syndrome to IBD are quietly reshaping how scientists think about type 2 diabetes.


Cardiometabolic Resilience: Who Avoids the Damage, and Why It Matters for You
Two new datasets reframe metabolic risk from inevitability to variability — hunting the protective phenotypes that shrug off vascular damage, and the routine labs that flag your five-year diabetes window.
Some people break the rules of cardiometabolic medicine. They carry the risk factors that are supposed to wreck an artery — three decades of Type 1 diabetes, a BMI that flags every actuarial table, kidneys failing toward transplant — and yet their vessels stay quiet. No calcification. No infarct. No story. For most of the last century, those people were statistical noise. A new Swedish protocol is treating them as the signal.
The shift matters for anyone in their late thirties or forties trying to make sense of a confusing risk picture. You eat well, you train, your labs look fine on paper — but your father had a stent at 52 and your fasting glucose has crept up two points a year. Are you on rails toward the same outcome, or is there real variability in how bodies handle metabolic stress? Two 2025 papers, taken together, suggest the answer is closer to variability than fatalism — and they hint at what to actually measure.
The resilience hunt
The first paper is the rationale and early results from the ESCAPER study, an exploratory project out of southern Sweden that started recruiting in September 2022. Rather than asking why high-risk patients get sick — the default question of cardiovascular medicine for a hundred years — ESCAPER asks the inverse: why don't the ones who should?
The cohort is built around three improbable groups. Type 1 diabetics with more than 30 years of disease and no macrovascular complications or macroalbuminuria. Obese adults with normal cardiac function who take no cardiovascular medications. And kidney-failure patients awaiting transplant whose arteries show no calcification. Each group is paired with controls and put through deep phenotyping: 24-hour blood pressure and ECG, vascular ultrasound, cardiac MRI, ergospirometry in a subgroup, plus biomarker and omics work. Kidney-failure participants also contribute arterial biopsies — actual vessel tissue, not just a blood draw.
The early read on 90 T1D patients and 31 obese participants is modest but telling: risk factors are well-managed, and the T1D subgroup posts a mean BMI of 25.6 — squarely in the normal range. That isn't the answer to the resilience question; it's the start of the question. The interesting biology lives in what the omics, imaging and biopsies reveal next.
For most of the last century, the people who broke the rules were statistical noise. A new protocol treats them as the signal.
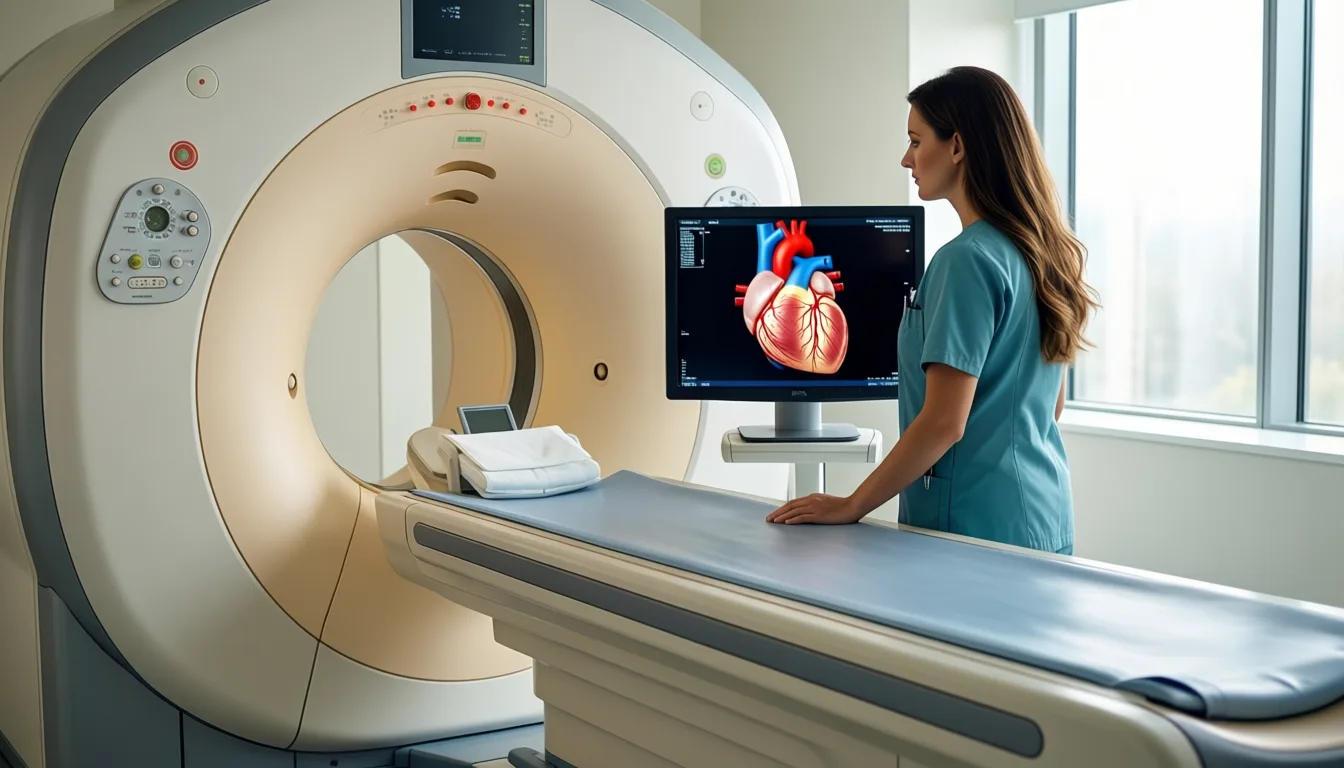
ESCAPER's deep phenotyping stack includes cardiac MRI, vascular ultrasound and ergospirometry — far beyond a standard physical.
What this changes for a 40-year-old
Practically? Not much yet. ESCAPER is an exploratory protocol; no protective mechanism has been confirmed, no drug target has been validated, and the early data are descriptive. What it does change is the framing of the conversation you have with your own physician. "High risk" is not a verdict. There are people with worse inputs than yours who never develop disease, and the field is finally trying to figure out why instead of treating them as outliers to be discarded.
The honest read on the evidence here is moderate. ESCAPER is a well-designed observational effort with strong phenotyping and Swedish registry follow-up — a real strength — but it has not yet produced mechanism-level findings, and observational work on resilience is famously prone to confounding. Translate the hype filter accordingly.
The other half: knowing your window
While ESCAPER asks who escapes, a second 2025 paper asks who's heading in. Researchers used Japanese health check-up data from 31,084 adults aged 30–69, collected between 2008 and 2016, to build prediction equations for five-year diabetes incidence. Participants with baseline diabetes or endocrine disease were excluded, and the population was split evenly into derivation and validation cohorts.
The headline result is unglamorous and useful. In the derivation cohort, five-year diabetes incidence was 5.0%. A model built from age, sex, body mass index, fasting blood glucose and HbA1c — five inputs you almost certainly already have on a recent lab panel — showed good discriminatory ability for predicting who would develop diabetes within five years. No proprietary biomarker. No genetic panel. Just the boring numbers from an annual physical, combined intelligently.
The trade-off worth naming: this is a Japanese cohort, and diabetes risk thresholds, body composition and incidence rates differ across populations. The equation's discriminatory power in a Swedish or American 40-year-old is not established. But the architectural lesson — that routine labs, modeled well, can define a personal five-year window — generalizes.

The Japanese prediction model relies on five inputs already on most annual lab panels.
How to use this — without overreaching
Two practical reframes come out of these papers for a busy 40-year-old optimizing energy, body composition and long-term metabolic health.
First, treat your fasting glucose and HbA1c as a trajectory, not a pass/fail line. The Japanese work suggests the combination of those two values, alongside age, sex and BMI, carries more predictive weight than any single number in isolation. If your HbA1c has drifted from 5.2 to 5.5 to 5.7 across three annual physicals while your BMI ticked up, the equation reads that pattern differently than a one-time 5.7 in a stable weight. Ask your clinician to look at the slope, not the snapshot.
Second, accept that resilience exists but don't bet on being resilient. ESCAPER is hunting for protective biology that may eventually become a drug, a screening test, or a phenotype you can actually identify in yourself. Today, none of that is in clinical use. The actionable layer remains the unglamorous one — sleep, training load, protein intake, alcohol, visceral fat — and the lab cadence to know which direction you're trending.
Neither paper licenses a supplement stack, a fasting protocol, or a specific intervention. Both quietly argue that the next decade of cardiometabolic medicine will be more personalized and less fatalistic than the last. That's the optimization story worth tracking.
- Resilience is real but not yet actionable. ESCAPER is mapping people who avoid vascular disease despite high-risk profiles; no protective mechanism is confirmed yet.
- Your annual labs already hold a five-year signal. A Japanese cohort of 31,084 built a validated diabetes prediction equation from age, sex, BMI, fasting glucose and HbA1c.
- Trajectory beats snapshot. Track glucose and HbA1c trends across years, not single readings.
- Evidence is moderate, not definitive. ESCAPER is exploratory; the Japanese equation is population-specific.
- The boring inputs still win. Sleep, training, body composition and lab cadence remain the levers — talk to a clinician about your personal risk window.
The framing has shifted. Cardiometabolic risk is variable, not fixed; some people genuinely escape damage that the standard models predict, and the routine labs in your file likely encode more about your personal window than you've been told. Neither finding is a green light to skip the cardiologist. Both are reasons to bring better questions to the appointment.
Frequently asked questions
What kinds of patients does the ESCAPER study recruit, and what makes them unusual?
ESCAPER recruits three groups: Type 1 diabetics with more than 30 years of disease and no macrovascular complications or macroalbuminuria, obese adults with normal cardiac function who take no cardiovascular medications, and kidney-failure patients awaiting transplant whose arteries show no calcification. Each group is paired with controls and put through deep phenotyping including cardiac MRI, vascular ultrasound, and biomarker and omics work.
What five data points does the Japanese diabetes prediction model rely on?
The model uses age, sex, body mass index, fasting blood glucose, and HbA1c — five inputs the article notes are already available on most annual lab panels, with no proprietary biomarker or genetic panel required.
Has ESCAPER confirmed a biological mechanism that explains why some high-risk people avoid cardiovascular disease?
No. The article states that no protective mechanism has been confirmed and no drug target has been validated; the early data are described as descriptive. ESCAPER is characterized as an exploratory protocol that has not yet produced mechanism-level findings.
Why might the Japanese prediction equation not apply directly to people from other countries?
The article notes that diabetes risk thresholds, body composition, and incidence rates differ across populations, so the equation's discriminatory power in a Swedish or American adult is not established. The authors describe this as a trade-off worth naming, while arguing that the broader lesson — that routine labs modeled together can define a personal five-year risk window — does generalize.
What does the article suggest is the practical takeaway for someone tracking their own metabolic health?
The article recommends treating fasting glucose and HbA1c as a trajectory rather than a pass/fail line, asking a clinician to look at the slope across multiple annual physicals rather than a single reading. It also advises against assuming personal resilience, since no protective phenotype is yet identifiable in clinical practice, and states that sleep, training load, protein intake, alcohol, and visceral fat remain the actionable levers.
Sources
- The ESCAPER study-exploring protective mechanisms against cardiovascular disease in subjects at high risk: rationale, study protocol, and first results. — Scandinavian cardiovascular journal : SCJ
- Development of risk prediction equations for 5-year diabetes incidence using Japanese health check-up data: a retrospective cohort study. — BMJ open

Sleep, Loneliness, and the Lifespan Connection
A new study of 2,297 adults suggests the variable your sleep tracker can't measure—how connected you feel—may be quietly shaping how you rest.
You can buy a ring that scores your REM, a mattress that cools to a tenth of a degree, and an app that nudges you to wind down at 9:47 p.m. What none of them measure is whether anyone is waiting up for you. A study published this year in Behavioral Sleep Medicine argues that may be a meaningful omission—because across 2,297 adults aged 19 to 99, the quality of a person's sleep and the loneliness they reported moved together, often closely, and in ways that shifted with age.
The paper, from Nielson, Boyle, and Dzierzewski, is not the first to suggest sleep and social life are entangled. It is, however, a careful attempt to map the relationship across the full adult lifespan using three validated instruments: the RU-SATED sleep health questionnaire, the Insomnia Severity Index, and the Gierveld Loneliness Scale, which separates emotional loneliness (missing a close confidant) from social loneliness (missing a wider network). The headline finding is straightforward: better sleep health tracked with lower loneliness, and worse insomnia symptoms tracked with higher loneliness, on both subscales and overall.
For a readership that treats sleep as an optimization problem—an input to tune, a variable to A/B test against caffeine cutoffs and magnesium glycinate—the implication is gentler than it sounds. The data are correlational. The study cannot tell you that calling a friend will fix your 3 a.m. wake-ups, nor that fixing your 3 a.m. wake-ups will make you feel less alone. What it can tell you is that the two tend to travel together, and that any serious account of why you are tired probably has to include who you are tired with.
- The association is real but correlational. Better sleep health and fewer insomnia symptoms tracked with lower loneliness across 2,297 adults, but the study cannot establish cause.
- Age shifts the picture. Younger adults reported lower loneliness; older adults reported higher loneliness, and age moderated how sleep and loneliness related.
- Loneliness has two flavors. Emotional loneliness (missing intimacy) and social loneliness (missing a network) were measured separately—and both moved with sleep.
- Your tracker is blind to half the equation. Wearables quantify sleep architecture; they do not capture the social context that may be shaping it.
- Talk to a clinician, not a forum. Persistent insomnia or loneliness are both treatable—and worth raising with a primary care provider.
What the study actually measured
RU-SATED is a six-item self-report that scores sleep on regularity, satisfaction, alertness, timing, efficiency, and duration—a broader frame than the single-number sleep score most wearables surface. The Insomnia Severity Index asks specifically about difficulty falling asleep, staying asleep, and the daytime consequences. The Gierveld scale, developed in the Netherlands and widely used in lifespan research, is the one that does the unusual work here: it treats loneliness not as a single feeling but as two related deficits, one intimate and one structural.
Participants—average age 44, spanning seven decades of adulthood—completed all three online. The authors then looked at direct associations and ran moderation analyses to ask whether the relationships between sleep and loneliness changed depending on how old the respondent was. Better sleep health and younger age were associated with lower loneliness on the total score and both subscales; greater insomnia symptoms and older age were associated with higher loneliness.

The study found loneliness rose with age—and that age changed how sleep and loneliness related to each other.
Any serious account of why you are tired probably has to include who you are tired with.
Why the age signal matters
The age effect is the part most likely to matter for how you think about your own sleep. Loneliness is not evenly distributed across a life; it tends to be higher at the ends—young adulthood and later life—and it interacts with sleep differently at different stages. The Nielson team's moderation analyses suggest the sleep–loneliness link is not a single equation that applies equally to a 28-year-old founder and a 78-year-old retiree.
For the executive demographic that tracks recovery scores obsessively, the practical reading is this: a stretch of poor sleep in your thirties or forties may be a signal worth interpreting socially, not only biologically. Travel, divorce, the slow attrition of friendships after a move—these are not the variables a wearable can ingest, but they are plausibly upstream of the metric it is showing you.
The wearable blind spot
The wellness-tech industry has spent a decade getting very good at measuring the body and very little time measuring the context the body lives in. A sleep score can tell you that your heart rate variability dipped and your deep sleep was short. It cannot tell you that you had a tense dinner, that your partner is traveling, or that the last person you spoke to in a non-transactional way was your barista.
The Nielson paper does not argue for adding a loneliness questionnaire to your morning app review. But it does sit within a growing body of work suggesting that sleep is not a purely physiological output—and that the variables we have made easy to measure may not be the ones doing the most work. For readers building a personal protocol around sleep, that is a useful corrective. Cooling pads and blackout curtains are real interventions. So, plausibly, is dinner with a friend.

Social contact is not a sleep intervention in any clinical sense—but the data suggest it is not unrelated either.
What to do with this, carefully
The honest answer is: not very much, prescriptively. The evidence is moderate, the design is cross-sectional, and the population is a single online sample. What the findings warrant is a small shift in framing. If you are tracking sleep and the numbers are stubbornly bad despite the obvious interventions—consistent schedule, dark room, no late caffeine, no late screens—it may be worth asking a less quantifiable question about the texture of your social week.
Persistent insomnia is a clinical condition with effective treatments, including cognitive behavioral therapy for insomnia (CBT-I), and it warrants a conversation with a primary care provider rather than another gadget. Persistent loneliness, similarly, is increasingly recognized as a health concern in its own right. Treating either as a personal failing to be optimized away tends to make both worse.
- If your sleep score is bad and you've tried the obvious fixes, consider the social variables your tracker can't see.
- Don't self-diagnose from a wrist sensor. Bring persistent insomnia to a clinician; CBT-I has strong evidence.
- Treat loneliness as health data. Two adults can have identical sleep architecture and very different recovery; context matters.
- Resist the urge to optimize a relationship. The intervention here is connection, not a protocol.
Frequently asked questions
What did the study find about the relationship between sleep and loneliness?
Across 2,297 adults aged 19 to 99, better sleep health tracked with lower loneliness, and worse insomnia symptoms tracked with higher loneliness on both subscales and overall. The association held for both emotional loneliness—missing a close confidant—and social loneliness—missing a wider network.
Does this mean that being lonely causes poor sleep, or that poor sleep causes loneliness?
The study cannot establish cause in either direction. It is a cross-sectional, self-report study, meaning the data are correlational and cannot tell you that fixing one condition will fix the other.
What is the difference between emotional loneliness and social loneliness as used in this research?
The Gierveld Loneliness Scale, the instrument used in the study, treats loneliness as two distinct deficits: emotional loneliness refers to missing a close confidant or intimate relationship, while social loneliness refers to missing a broader social network. Both types moved alongside sleep measures in the findings.
How does age factor into the sleep-loneliness connection?
Age moderated the relationship, meaning the link between sleep and loneliness did not apply equally across all life stages. Younger adults reported lower loneliness while older adults reported higher loneliness, and the study's moderation analyses suggest the sleep-loneliness association shifts depending on how old the respondent was.
Why can't a wearable sleep tracker fully explain why someone is sleeping poorly?
According to the article, wearables quantify sleep architecture—metrics like heart rate variability or deep sleep duration—but cannot capture the social context shaping that sleep, such as relationship stress, isolation, or the quality of a person's social week. The article describes this as a meaningful blind spot in how wellness technology measures sleep.
Sources

Rewiring Glucose Control: What Three New Papers Say About Gut, Liver, and Diabetes
Microbiota-targeted therapies, a newly mapped liver enzyme called HYAL1, and Mendelian evidence linking metabolic syndrome to IBD are quietly reshaping how scientists think about type 2 diabetes.
For decades, type 2 diabetes has been told as a story about two organs and one hormone: a pancreas that can't keep up, muscles and liver that stop listening, and insulin caught in the middle. That story isn't wrong — it's just getting crowded. A wave of new research is widening the cast to include the trillions of microbes in the gut, a little-known enzyme in the liver, and a surprising genetic thread that ties metabolic syndrome to inflammatory bowel disease. None of it is ready for the pharmacy shelf. All of it is worth paying attention to.
If you've spent any time on health TikTok this year, you've heard people talk about glucose spikes the way they used to talk about cortisol. The science underneath that conversation is genuinely shifting — but slowly, and in directions that don't always match the captions. Three recent papers, taken together, sketch a more interesting picture: type 2 diabetes as a whole-body conversation between the gut, the liver, and the immune system, rather than a single broken switch.
The first thread is the gut microbiome. A 2025 review in La Tunisie Medicale takes stock of a fast-moving field: people with type 2 diabetes consistently show a pattern of gut dysbiosis — a shifted balance of bacterial species — and researchers are now testing whether nudging that balance back can help correct the underlying insulin resistance. The review surveys strategies from prebiotics and probiotics to fecal microbiota transplantation, framed as a complement to (not a replacement for) the standard trio of medication, movement, and a lower-carbohydrate diet.
- Three separate findings, one direction: type 2 diabetes biology is looking less like a single broken pathway and more like a network problem across gut, liver, and immune system.
- Microbiome therapies are promising but unproven: a 2025 review catalogues the strategies; it does not declare any of them ready for routine clinical use.
- A new liver enzyme enters the chat: HYAL1 helps the liver stop making glucose after a meal — so far, shown in mice.
- Metabolic syndrome and IBD may share a causal thread: Mendelian-randomization analysis suggests waist circumference and hypertension are linked to higher risk of Crohn's and ulcerative colitis, respectively.
- None of this changes today's care: these are early signals, not prescriptions. Talk to a clinician before changing anything.
The gut: dysbiosis as a target, not just a symptom
The microbiome story in metabolic disease has been simmering for years. What's new in the recent literature is the framing: researchers are increasingly treating dysbiosis as something to act on, not just measure. The La Tunisie Medicale review summarises the current toolkit for modulating the gut microbiota to correct dysbiosis in people with type 2 diabetes, drawing on advances in metagenomics and metabolomics that finally let scientists see, at species-level resolution, what's actually shifting.
It's worth being honest about what a review like this can and can't tell us. It catalogues directions, not verdicts. The authors describe a research frontier where probiotics, prebiotics, synbiotics, dietary fibre interventions, and fecal microbiota transplantation are all being tested as ways to dent insulin resistance — but none of these are positioned as a stand-alone replacement for established care. If your feed is selling you a single capsule that fixes blood sugar, the literature is not where that confidence comes from.

Fermented foods and fibre dominate the lifestyle conversation around the microbiome — but the clinical evidence for reversing diabetic dysbiosis is still emerging.
The liver: meet HYAL1, the postprandial brake
The second thread is, in some ways, the most mechanistically satisfying. One of the defining features of type 2 diabetes is that the liver doesn't quite get the memo when you eat. Normally, after a meal, hepatic gluconeogenesis — the liver's ability to manufacture glucose from non-carbohydrate sources — should switch off, letting insulin route incoming nutrients into glycogen and fat for storage. In insulin resistance, that switch sticks.
A 2025 study in Life Metabolism proposes a new player in that switch: hyaluronidase-1, or HYAL1, a lysosomal enzyme that breaks down circulating hyaluronan after meals. In mice lacking HYAL1, gluconeogenesis stayed inappropriately switched on; in mice with extra HYAL1 in the liver, gluconeogenic activity went down, and the metabolic chaos triggered by a high-fat diet was blunted. Mechanistically, the authors trace a path through the cell's UDP-GlcNAc pool and a modification called O-GlcNAcylation, ending at the mitochondria — where less ATP synthase modification means less ATP, and less fuel for making new glucose.
That's a lot of biochemistry for a magazine paragraph, and the honest translation is this: HYAL1 looks like a previously unappreciated brake on the liver's after-meal glucose output. The asterisk, also honest: this work was done in mice. Mouse liver biology is informative, not destiny. Whether HYAL1 becomes a future drug target, a biomarker, or a footnote depends on work that hasn't been done yet.
HYAL1 looks like a previously unappreciated brake on the liver's after-meal glucose output — in mice. That's a beginning, not a verdict.
The immune angle: a Mendelian link to IBD
The third paper takes the widest lens. Epidemiologists have noticed that metabolic syndrome and inflammatory bowel disease — Crohn's and ulcerative colitis — have been rising in parallel, and that people with both tend to do worse. Correlation isn't causation, but a study technique called Mendelian randomization can get closer to causation by using genetic variants as natural experiments.
A bidirectional two-sample Mendelian-randomization analysis published in Archives of Medical Science did exactly that, in European population data. The headline finding: genetically predicted metabolic syndrome was associated with higher risk of Crohn's disease (OR 1.34, 95% CI 1.009–1.779). Drilling into components, waist circumference was linked to higher Crohn's risk (OR 1.33), and hypertension to higher ulcerative colitis risk (OR 1.61). In the reverse direction, IBD appeared to nudge triglyceride levels up, modestly.
What this is: suggestive evidence that the metabolic and the inflammatory aren't separate countries. What it isn't: a reason to assume a flat stomach prevents Crohn's, or that managing blood pressure will spare you ulcerative colitis. Mendelian randomization is powerful, but it's a statistical lens on populations, not a forecast for any individual reader.

The practical message hasn't changed: sleep, movement, fibre, and regular check-ins with a clinician still do most of the heavy lifting.
What this means if you're not a researcher
It is tempting, when three papers point in interesting new directions, to turn that into a shopping list — a probiotic to buy, a supplement to chase, a number to optimise. Resist. The most accurate read on this body of work is that the map of type 2 diabetes is getting more detailed, not that the route has changed.
The microbiome work is real but still preliminary in humans. The HYAL1 finding is elegant but lives, for now, in mice. The Mendelian-randomization signal is intriguing but statistical. Each one is a thread; none is yet a rope you can hang treatment on. If you have type 2 diabetes, prediabetes, metabolic syndrome, or IBD — or you're worried you might — the right next step is a conversation with a clinician who knows your history, not a re-org of your supplement drawer.
The reason these papers are worth reading together isn't that any one of them rewrites the textbook. It's that, taken as a set, they point toward a version of type 2 diabetes that's less about a single failed organ and more about a conversation — between gut microbes, hepatic enzymes, and immune signalling — that can go off-key in many places at once. That's a more complicated story. It's also, eventually, a more hopeful one, because a network has more places to intervene than a single broken switch.
For now, the assignment is unglamorous and unchanged: sleep, move, eat enough fibre to make your gut bacteria happy, see your doctor, and treat your feed's certainty with affectionate skepticism. The science is moving. The hype is moving faster. The gap between them is where careful readers live.
Frequently asked questions
What is dysbiosis, and why do researchers think it matters for type 2 diabetes?
Dysbiosis refers to a shifted balance of bacterial species in the gut. Researchers are increasingly treating it as something to act on rather than just measure, because people with type 2 diabetes consistently show this pattern, and studies are testing whether correcting it can help address the underlying insulin resistance.
What is HYAL1, and what role might it play in blood sugar regulation?
HYAL1, or hyaluronidase-1, is a lysosomal enzyme in the liver that breaks down circulating hyaluronan after meals. Research in mice suggests it acts as a brake on the liver's after-meal glucose production, with mice lacking HYAL1 showing inappropriately elevated gluconeogenesis and those with extra HYAL1 showing reduced gluconeogenic activity.
Was the HYAL1 research conducted in humans?
No. The HYAL1 findings come from mouse studies. The article notes that mouse liver biology is informative but not destiny, and whether HYAL1 becomes a drug target, a biomarker, or a footnote depends on research that has not yet been done.
What did the Mendelian randomization study find about the link between metabolic syndrome and inflammatory bowel disease?
The analysis found that genetically predicted metabolic syndrome was associated with a higher risk of Crohn's disease, with waist circumference specifically linked to Crohn's risk and hypertension linked to higher ulcerative colitis risk. In the reverse direction, IBD appeared to modestly raise triglyceride levels.
Do any of these three research findings change what people with type 2 diabetes should do today?
According to the article, none of these findings change current care — they are described as early signals, not prescriptions. The article states that sleep, movement, fibre, and regular check-ins with a clinician still do most of the heavy lifting, and advises talking to a clinician before changing anything.
Sources
- New therapeutic approaches based on modulation of the intestinal microbiota to correct dysbiosis in patients with type 2 diabetes. — La Tunisie medicale
- Hyaluronidase-1 mediates postprandial suppression of hepatic gluconeogenesis. — Life metabolism
- Association between metabolic syndrome and inflammatory bowel disease: a bidirectional two-sample Mendelian randomized study. — Archives of medical science : AMS